Through the literature review, we found that Aca1 protein was important in the survival of Pseudomonas phages1. Such protection depends on the interaction between Aca1 protein and a promoter (acr-promoter) that regulates the expression of acr gene1. Therefore, we developed an idea that we could disrupt the binding between Aca1 protein and acr-promoter to induce the death of Pseudomonas phages. We expected such disruption was carried out by a drug, a small molecule. To develop such a drug, we designed a screening system to firstly find a potential inhibitor and then optimize it. Such a system was built in our experiment.
Engineering Success
how we succeed in the engineering
Overall Engineering Success
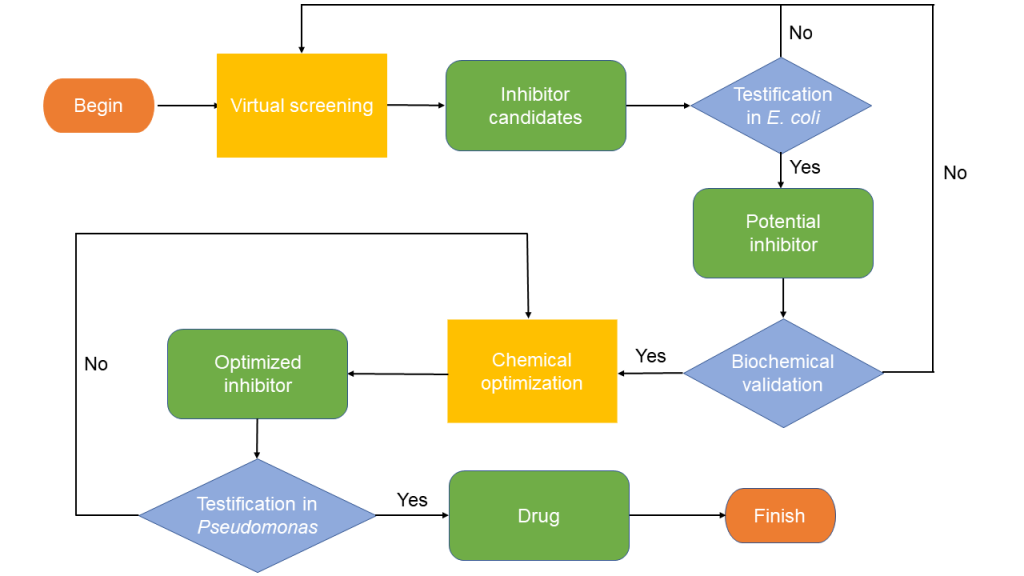
Figure 1. The workflow of our screening system
In this screening system, we firstly screened inhibitor candidates in our virtual library. And then, for those inhibitor candidates, we tested if they were functional as our expectation in our cellular screening system built-in E. coli. For the ones that resulted in we expect results, we were going to further demonstrate their binding with Aca1 via biochemical approaches in order to avoid false positives. As such potential inhibitors may not reach the highest efficiency, after biochemical validation we planned to optimize them via chemical access. For those optimized inhibitors, they would be tested in Pseudomonas to find out if it could really defend against the infection of phages. Finally, the ones that succeeded in defending phage infection of Pseudomonas were the drugs we wanted.
Engineering Success in E. coli validation system
In our E. coli validation system, we expected to successfully screen our inhibitor candidates with a system simulating phage infected Pseudomonas. In such a system, we could screen for effective inhibitors for the binding between Aca1 and acr-promoter. We got an idea to use the expression level of eGFP2, a fluorescence protein, to reflect the activity of acr-promoter. That is to say, in our system, a stronger fluorescence illuminance could reflect a higher activity of acr-promoter. As Aca1 protein could bind to acr-promoter and inactivate it1, we expected a decrease in fluorescence illuminance with the presence of Aca1 protein and recovery of fluorescence illuminance after an effective inhibitor was added.
Therefore, the challenge we faced in building such a validation system was how to design functional plasmid to realize a simulation system in E. coli. We wanted to achieve a successful system by designing plasmids. Then, we were going to test the feasibility of such a plasmid system for the expected lowering of fluorescence illuminance in the presence of Aca1. If it turned out that such a plasmid system worked, we would then use such a system to screen our inhibitor candidates with the model we built. Finally, the one that turned out to have the highest efficiency was what we wanted.
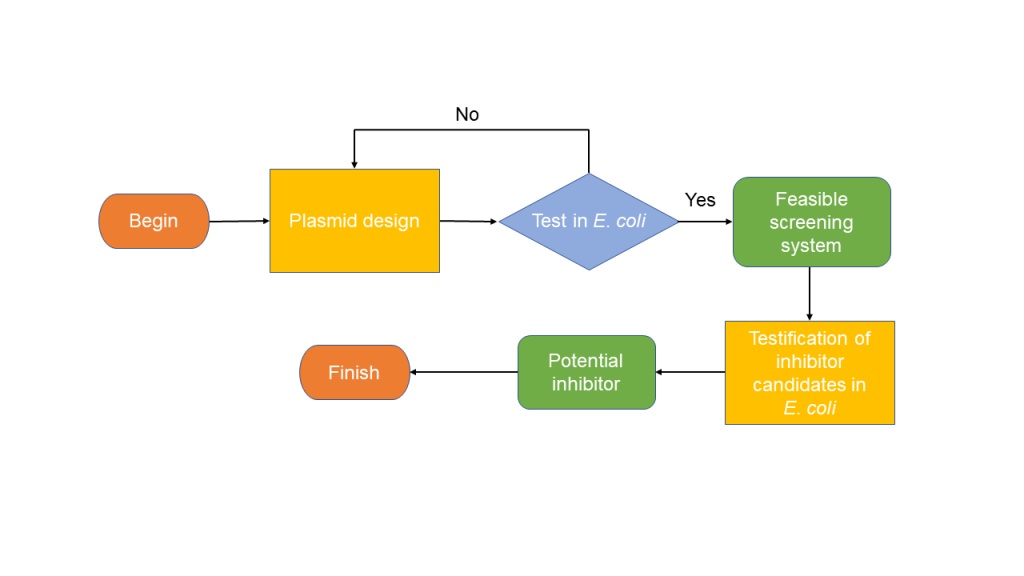
Figure 2. The workflow of building E. coli validation system
In the first attempt, we build a plasmid that was expected to carry all the important information in our expected system. On this plasmid, acr-promoter regulated the transcription of egfp gene. And aca1 gene was also located on this plasmid under the regulation of lacI promoter to ensure a steady high transcription level. However, after transforming such plasmid into E. coli, we found that the fluorescence illuminance in such E. coli is the same as the E. coli transformed with a plasmid without any aca1 gene. Such results indicated that the aca1 gene in such a system failed to be expressed. Therefore, this system could not work as our expectation and we tried another way to design plasmids.
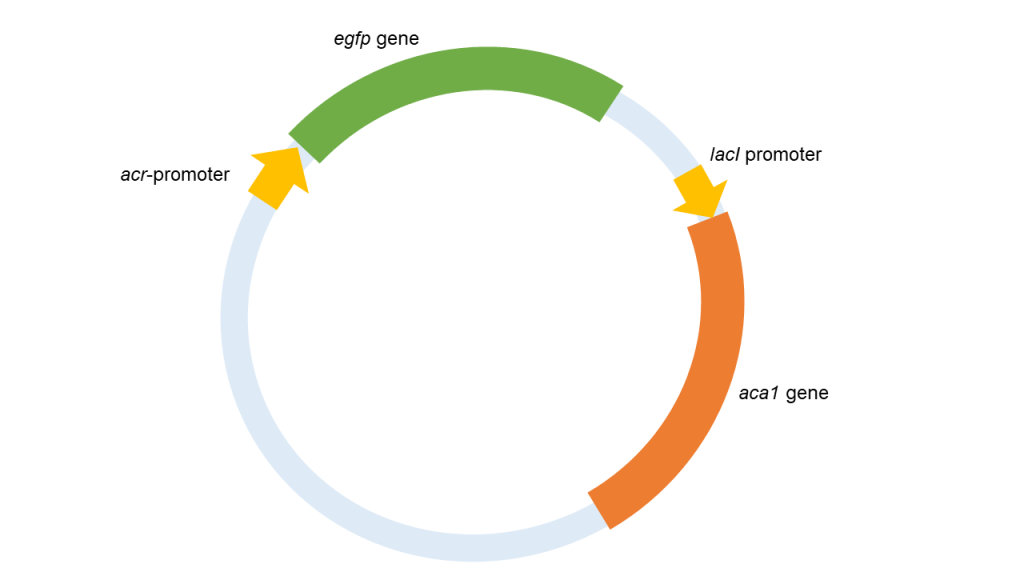
Figure 3. Illustration of plasmid design at the first attempt
In the second attempt, we turned to create a two-plasmid system, one plasmid for Aca1 expression and the other one for reporting. In this way, we engineered the plasmid pUCP20 (Pseudomonas plasmid) by inserting the report gene egfp after the acr-promoter. And aca1 gene was inserted after lac operon in the plasmid pRSFduet. Then, the engineering pUPC20 and pRSF1b plasmids were co-transformed into E. coli (BL21DE3). Due to the different antibiotic-resistant genes the two plasmids had, we readily selected the E. coli which successfully co-transformed by using ampicillin and kanamycin. And we found that such a system just worked as our expectation.
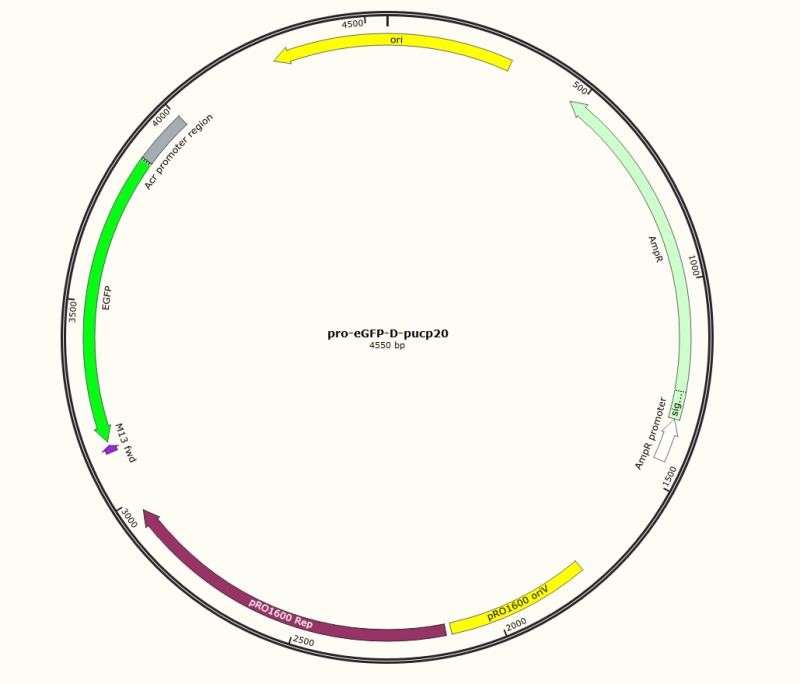
Figure 4. Engineering pUCP20 plasmid
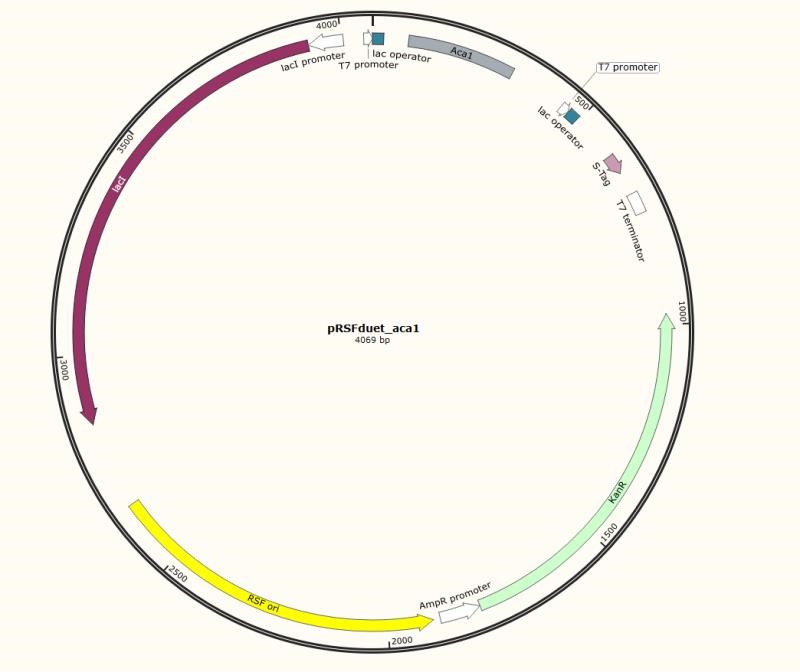
Figure 5. Engineering pRSFduet plasmid
Finally, using the two-plasmid system (a composite Part: BBa_K3423005 made up of BBa_E0040, BBa_J61104, BBa_I719005, BBa_B0012, BBa_K3423000, and BBa_K3423001)we developed at our second attempt, we succeeded in screening potential inhibitors among our inhibitor candidates with our model.
Table 1. in vivo KI of different PDI candidates
| compounds | c(PDI-inhibitor) (μM) | I0 | I | I1 | KI (μM) |
|---|---|---|---|---|---|
| 1388-F10 | 19 | 167401 | 191357 | 1446723 | 115.2079 |
| 1388-B9 | 19 | 167401 | 192753 | 1446723 | 108.743 |
| 1401-H11 | 19 | 167401 | 220510.3 | 1446723 | 50.75995 |
| 1401-G2 | 19 | 167401 | 222787 | 1446723 | 48.58307 |
| 1402-C10 | 19 | 167401 | 227404.7 | 1446723 | 44.6751 |
| 1388-D4 | 19 | 167401 | 228061.7 | 1446723 | 44.16743 |
| 1388-H6 | 19 | 167401 | 237461.3 | 1446723 | 37.94673 |
| 1405-H10 | 19 | 167401 | 271971.7 | 1446723 | 24.69803 |
| 1389-B10 | 19 | 167401 | 87026.33 | 1446723 | -37.192 |
| 1400-C4 | 19 | 167401 | 146922 | 1446723 | -139.539 |
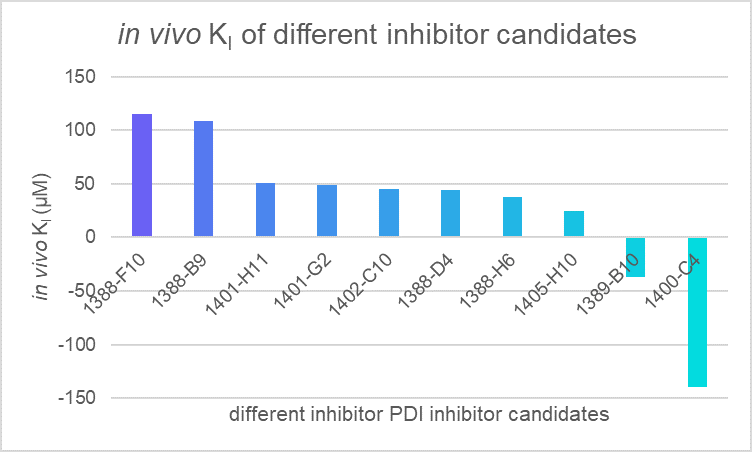
Figure 6. in vivo KI of different PDI candidates
References
- Stanley, S. Y., et al. Anti-CRISPR-Associated Proteins Are Crucial Repressors of Anti-CRISPR Transcription. Cell 178, 1452-1464.e1413, doi:https://doi.org/10.1016/j.cell.2019.07.046 (2019).
- Zhang, G., Gurtu, V. & Kain, S. R. An Enhanced Green Fluorescent Protein Allows Sensitive Detection of Gene Transfer in Mammalian Cells. Biochemical and Biophysical Research Communications 227, 707-711, doi:10.1006/bbrc.1996.1573 (1996).