Large-Scale fermentation using a densely-cultured bacterium that is utilized in industrial production often suffer severely from bacteriophage infection due to the lack of genetic diversity. Phage contamination is a worldwide problem in the fermentation industry. Here we present a chemical biology approach to developing a compound that can protect Pseudomonas from phages infection.
Results
Wow~That’s amazing~
Overview
Protein-ligand Docking
In silico screening was performed based on the Aca1 structure solved by our primary PI using the software Molecular Operating Environment (MOE) and several promising candidates were identified.
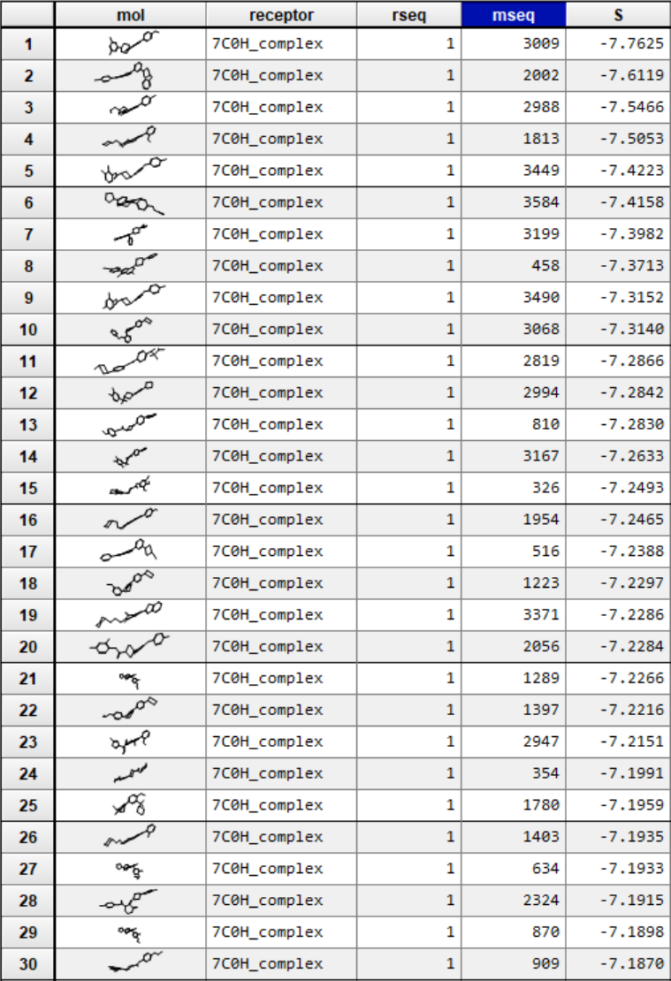
Table 1. In silico screening results
Report system constructing and in vivo screening
In the first plasmid, Aca1 is expressed by a promoter regulated by LacI. In the second, green fluorescent protein (GFP) is expressed by the Acr promoter, which is inhibited by Aca1 expressed by the first plasmid.
As when the critical binding site of protein Aca1 (the 44th amino acid) is mutated, Aca1 can no longer bind to the target DNA. We mutated the aca1 gene in the first plasmid (the 44th amino acid arginine is mutated to alanine) as a positive control.
With the presence of 500uM IPTG, Aca1 is overexpressed by the first plasmid to inhibit the acr promoter in the second plasmid. Thus, the expression of GFP inhibited. While for the positive control, as Aca1 is mutated to Aca1R44A, the acr promoter is not inhibited, and thus the expression level of GFP is higher than that of the wild type (WT) control. Though detecting the green fluorescence intensity, we can easily measure the expression level of the GFP and thus determine at what level acr promoter is inhibited. Due to the inhibition from Aca1, the green fluorescent intensity difference can be over 10 times, which proved the efficiency of this reporting system.
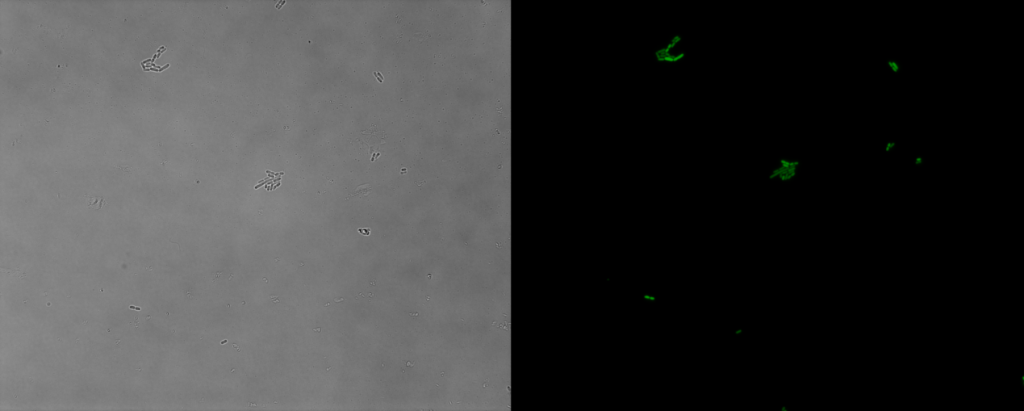
Figure 1. The green fluorescent protein expressed by acr promoter in E. coli
We can detect green fluorescence from almost every E. coli cell, which means the transformation is successful and the report system is effective.
Then, 30 candidates we got form the virtual screening is tested, we applied our model to evaluate the efficiency of 10 different PDI inhibitor candidates and found that 1400-C4 has the smallest in vivo KI among those candidates. Therefore, we concluded that 1400-C4 could have the best inhibition efficiency and focused on 1400-C4 in the further experiment process.
Table 2. in vivo KI of different PDI candidates
| compounds | c(PDI-inhibitor) (μM) | I0 | I | I1 | KI (μM) |
|---|---|---|---|---|---|
| 1388-F10 | 19 | 167401 | 191357 | 1446723 | 115.2079 |
| 1388-B9 | 19 | 167401 | 192753 | 1446723 | 108.743 |
| 1401-H11 | 19 | 167401 | 220510.3 | 1446723 | 50.75995 |
| 1401-G2 | 19 | 167401 | 222787 | 1446723 | 48.58307 |
| 1402-C10 | 19 | 167401 | 227404.7 | 1446723 | 44.6751 |
| 1388-D4 | 19 | 167401 | 228061.7 | 1446723 | 44.16743 |
| 1388-H6 | 19 | 167401 | 237461.3 | 1446723 | 37.94673 |
| 1405-H10 | 19 | 167401 | 271971.7 | 1446723 | 24.69803 |
| 1389-B10 | 19 | 167401 | 87026.33 | 1446723 | -37.192 |
| 1400-C4 | 19 | 167401 | 146922 | 1446723 | -139.539 |
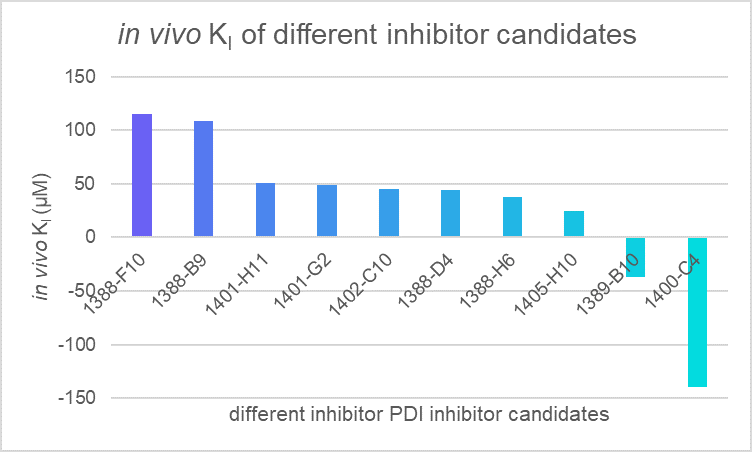
Figure 2. in vivo KI of different PDI candidates
Then, we repeated several experiments using 1400-C4:
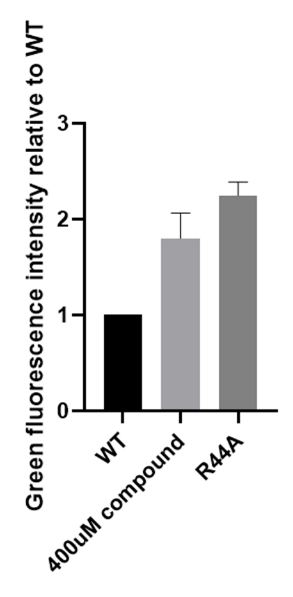
Figure 3. Efficiency of Navitoclax measured with green fluorescence intensity using part BBa_K3423005
Confirm the binding with differential scanning fluorimetry (DSF)
We confirmed the binding of the compound and Aca1 in vitro through DSF:
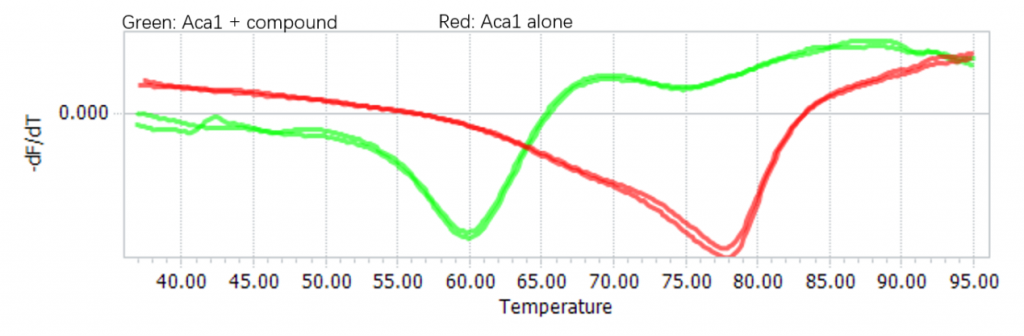
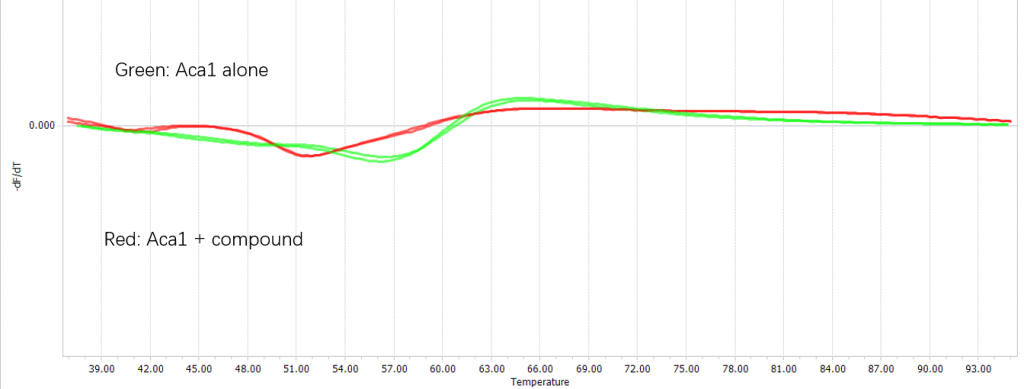
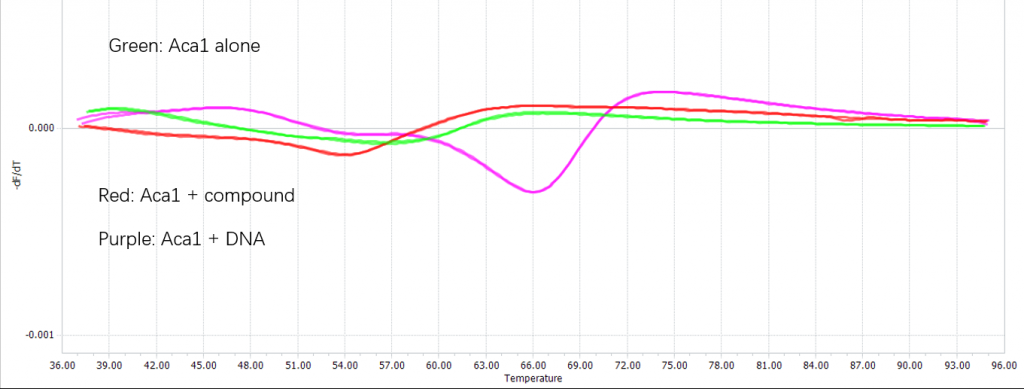
Figure 4. Binding between Navitoclax and Aca1 confirmed using DSF
Compound Design and Hit Optimization
In our previous work, we got a good result through a long period of time of virtual screening. Fortunately, the results of biological experiments show that the molecule has a certain regulatory effect on the protein aca1. However, the computer can only give us a rough calculation result based on the analysis of the volume of the protein pocket, and the molecule obtained does not show excellent performance in the interaction with the protein. Therefore, we need to analyze the molecular docking model, to optimize the structure of the hit, in order to improve its biological activity.
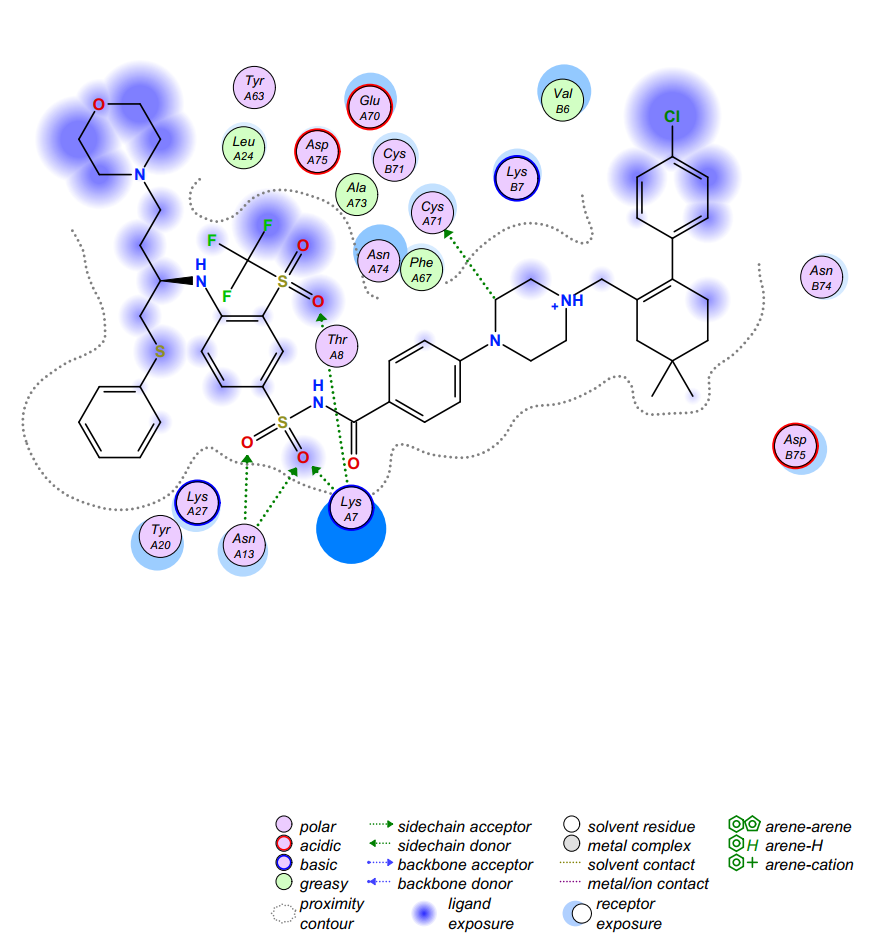
Figure 5. Computer virtual molecular docking model of Navitoclax (in black) in complex with the aca1. Dotted arrows indicate interactions, and the spheres denote the surrounding amino acids.
We have obtained a hit compound through virtual screening and the docking model of it with aca1 protein is shown in Figure 5. Through the analysis of the model, we can divide the structure of Navitoclax into five regions according to the binding strength of the structural fragment with the residue amino acids and the spatial structure of the protein pocket. The five regions are shown in Figure 6, distinguished by different colors.
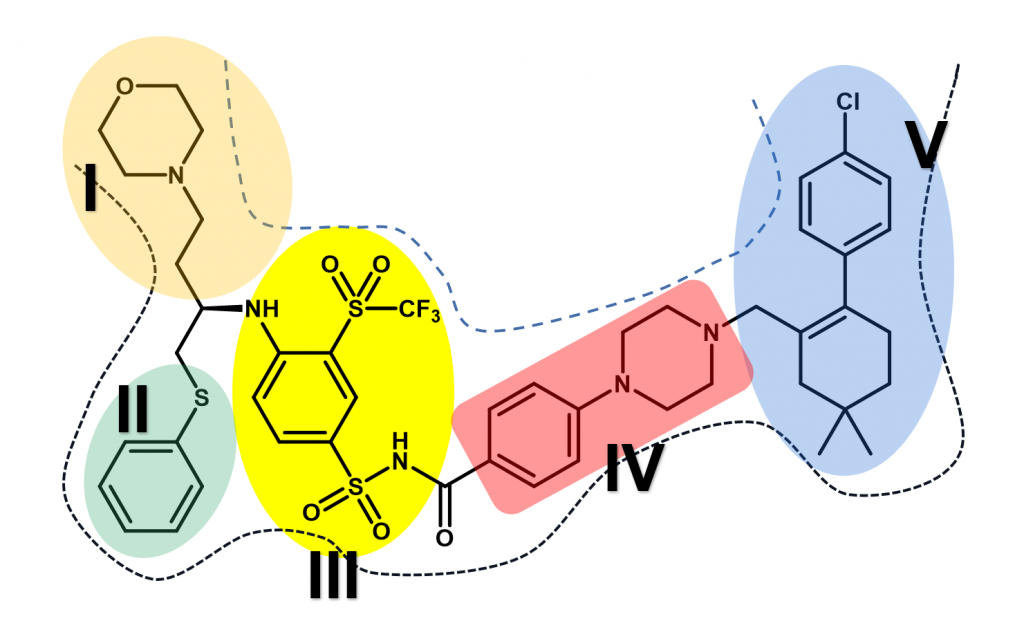
Figure 6. Five regions of Navitoclax. Dotted lines indicate the inner surface of the aca1 protein, and the five regions are distinguished with different colors.
According to the model, we found that two regions in the molecular structure should be retained in the subsequent molecular design and optimization. One of them is the structural segments of sulfonamides in Navitoclax (region III) because it plays an important role and exhibits strong interactions with LysA7 and AsnA13. We have reason to believe that this fragment may be a necessary structure for Navitoclax to be active in aca1 protein. The other is the structure of morpholine (region I), which places in the solvent region of the pocket. Based on our understanding of the solvent region, there should be a water-soluble functional group to improve the binding ability with protein and improve the water solubility of the molecule. Morpholine, a highly water-soluble functional group can do this job excellently. So, this kind of structure will be retained in the following design and optimization.
However, another solvent region is occupied by a fat-soluble aromatic ring (region V). Moreover, no amino acid residue can interact with the aromatic ring, which is an unnecessary fragment. What’s more, the site has brought us some difficulties in chemical synthesis, which is not cost-effective in terms of economic benefits. Considering various factors, we decided to delete the aromatic ring above region V and modify the group below. We found that there is an acidic amino acid residue AspB75 at the bottom of region V. If we add an alkaline ring to this part, we have reason to believe that there will be a strong interaction between them. The common basic structure includes a large number of aromatic nitrogen heterocycles, saturated nitrogen heterocycles, and simple amine groups. But finally, we decided to use imidazole and triazole because they have the advantages of easy synthesis, moderate alkalinity, suitable size, proper rigidity, and so on.
Similarly, there is also a meaningless fragment in Region II, the thioether, which increases the cost of its synthesis but shows no significant interaction between the residue amino acids. As a result, we can replace the sulfur atom with a simple carbon atom. Then, considering there is an exposed basic amino acid residue LysA27 closed to Region II, an acidic group is used to replace the aromatic ring below to generate interaction with it. Generally speaking, the structure of carboxylic acid is a good choice. However, to enhance the membrane permeability of molecules, we can use the principle of bioisosteres to replace the structure of carboxylic acid with the structure of tetrazole.
In addition to analyzing the binding effect, we can also optimize our molecular structure by analyzing the spatial structure of the protein pocket. The optimization of region IV is a good example. Through the analysis of the spatial structure, we know that the space occupied by Region IV is not too wide. Therefore, we can choose to replace the ring structure of the region with an ordinary carbon chain or replace it with smaller rings. If we replace it with an ordinary fatty chain, the resulting molecule is likely to lose its rigidity and thus fail to perform the expected interaction well. Considering the above factors, we try to replace the region with an alkyne structure and five-membered heterocycles. At the same time, we have to consider whether we can optimize region V from the combination ability. We found that there are an amino acid residue CysA71 and PheA67 above region four, so we can use a structure of β-unsaturated carbonyl structure in region four to produce an n → π* type of interaction with CysA71 or π→ π*type of interaction with PheA67.
Through the theoretical analysis of the molecular docking model, we designed the second generation of small molecules to regulate Aca1 protein, and their structure and docking results are as follows.
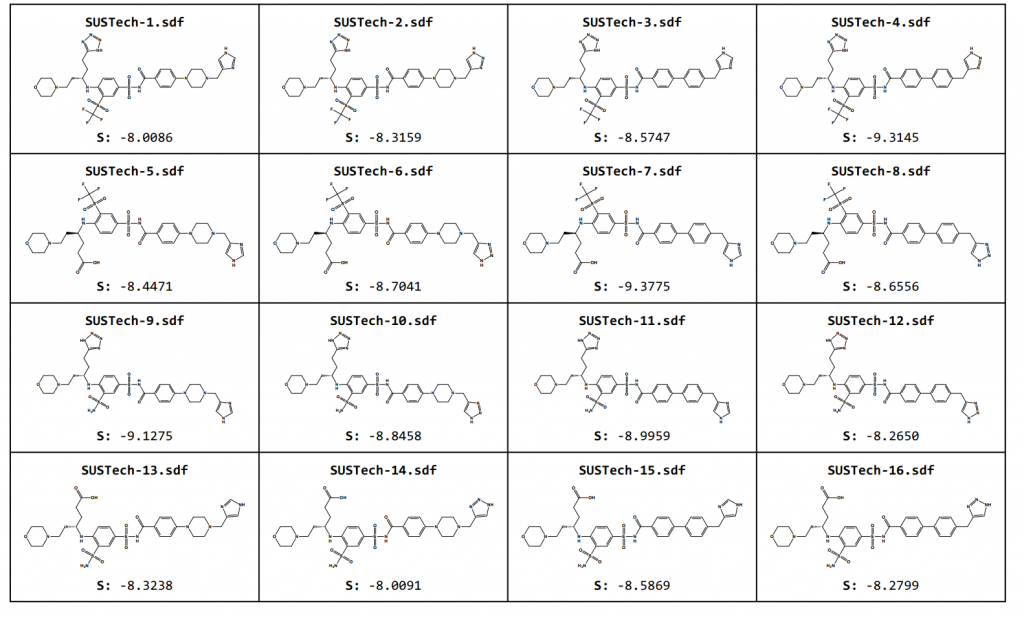
Figure 7. Optimized Aca1 inhibitors based on Navitoclax
Our second-generation molecules, in the design of regions I and II, chose to retain the same length of carbon chain as the first generation (Navitoclax). However, in the subsequent retrosynthesis analysis, we found that if we need to synthesize such a structural fragment (regions I and II), we need to use a polarity reversal dipole, such as α-halocarbonyl compound or cyanogen ion. Because α-halocarbonyl compounds have some problems in selectivity, and cyanogen compounds are highly toxic substances, which are rarely used in industry, even in the laboratory, which makes our synthesis and industrial synthesis very difficult.
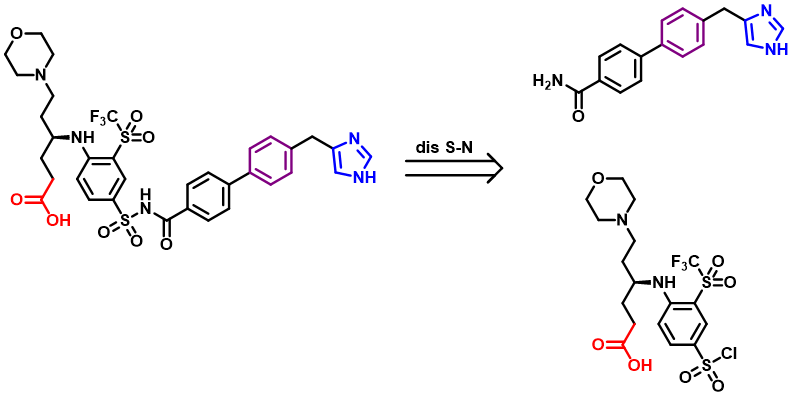
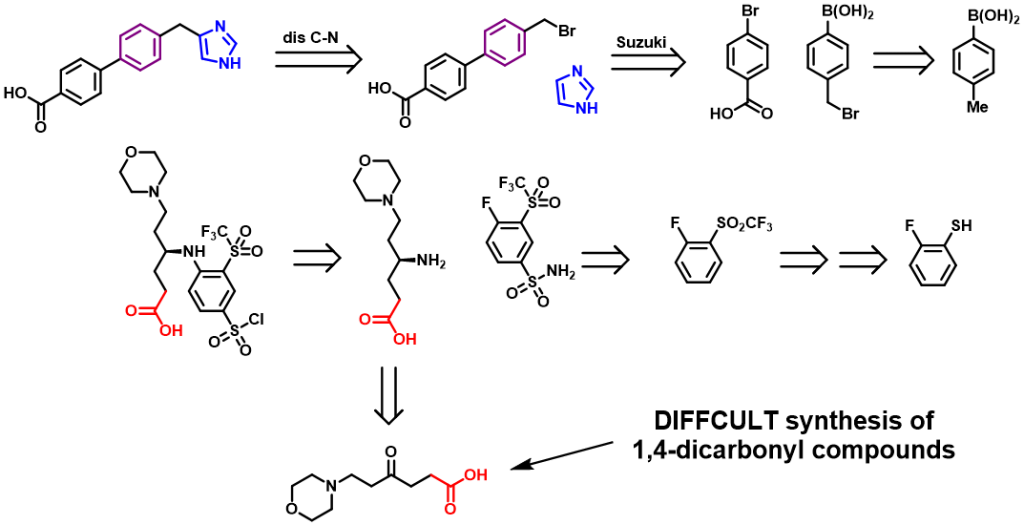
Figure 8. Retrosynthesis of one Aca1 inhibitor
Therefore, we further designed the third-generation molecule to regulate Aca1 protein. The third-generation molecules further enrich the functional groups of molecular structure, so as to maximize the binding strength of molecules and proteins, so as to reduce the amount of actual use and reduce the cost. At the same time, we hope to optimize the physical and chemical properties of the molecules, such as water solubility. What’s more, we abandoned the structure fragments that are difficult to synthesize in the second-generation molecules, hoping to reduce the difficulty of synthesis and production costs. And their structures are shown below:
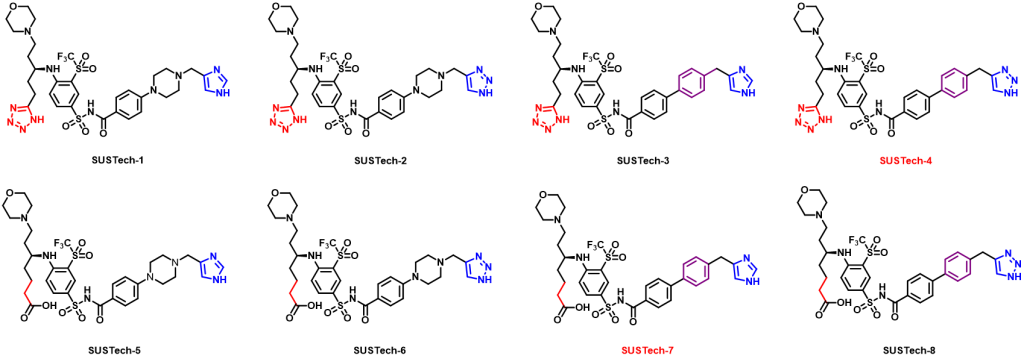
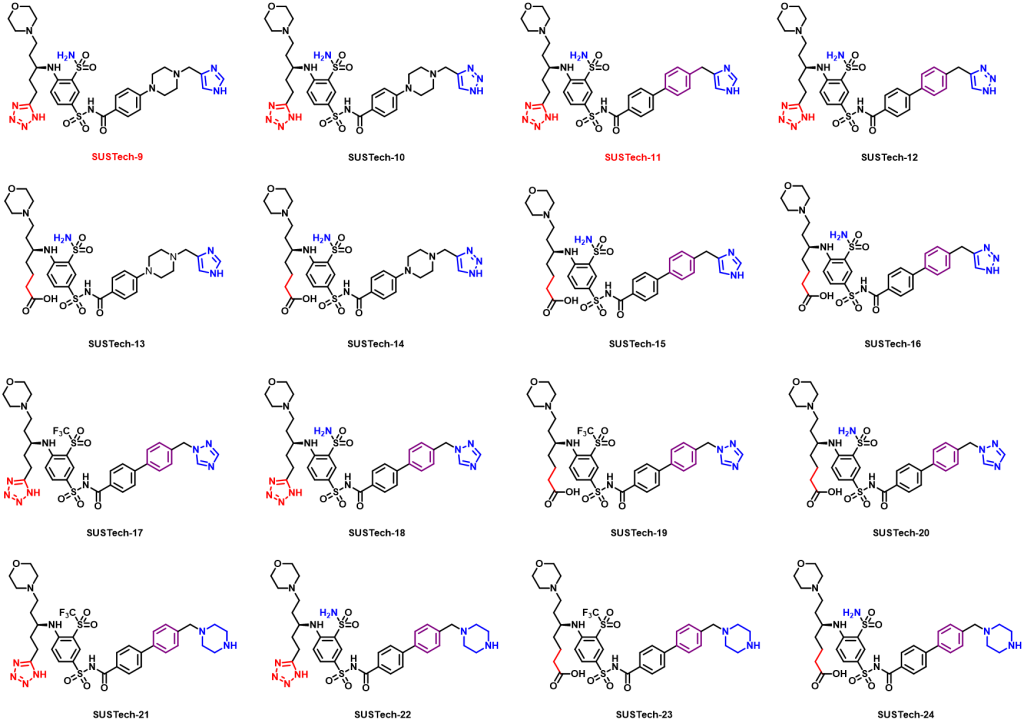

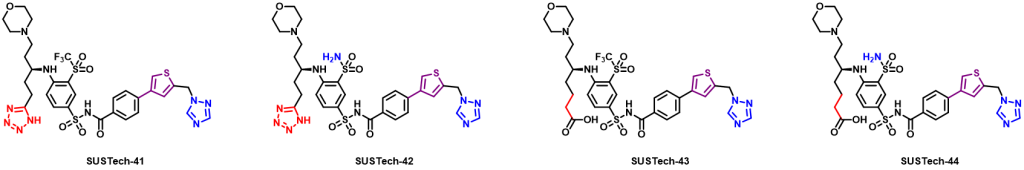
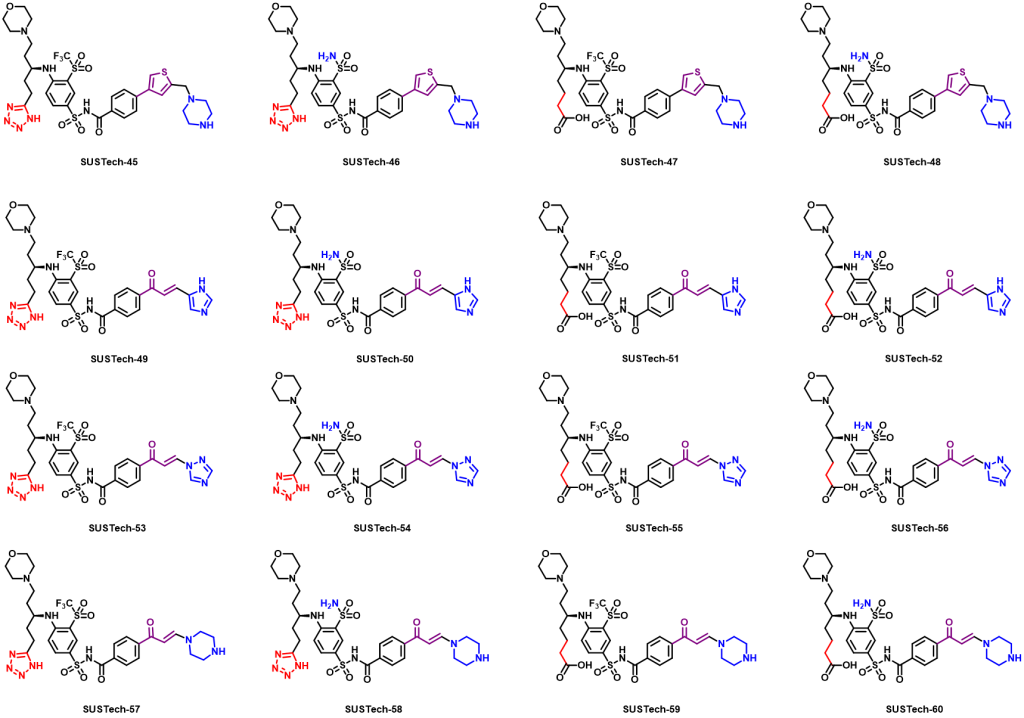
Figure 9. Aca1 inhibitors (third generation)